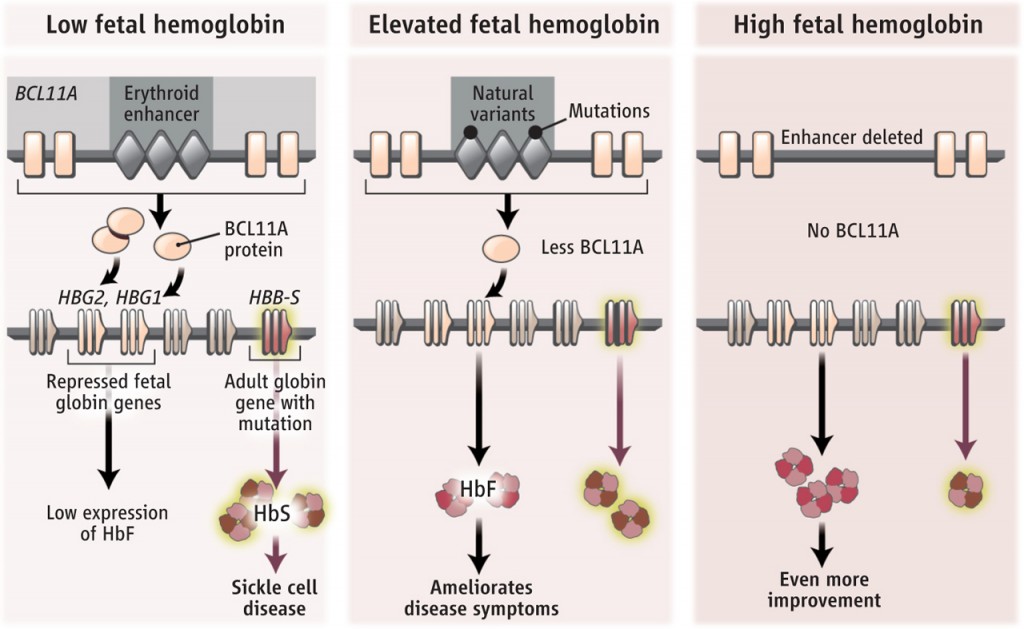
Los trastornos del gen de la βglobina, como la enfermedad de células falciformes (SCD) es el resultado de mutaciones genéticas en el gen de la β-globina, y fue uno de los primeros trastornos abordados con terapia específica epigenética. Las estrategias para la terapia epigenética se centraron en la reactivación de la expresión fetal de γ-globinaa.
El curso clínico de las β-hemoglobinopatías se ve mejorado por los niveles elevados de γ-globina fetal, que reduce el desequilibrio de la cadena de globina en las β-talasemias y ejerce un potente efecto antisickling en SCD. Las deleciones grandes que ocurren naturalmente que abarcan los genes de globina β producen un aumento congénito de la expresión de hemoglobina fetal (HbF) conocida como persistencia hereditaria de HbF (HPFH), que mejora los fenotipos SCD. Se cree que las grandes deleciones de HPFH eliminan las secuencias inhibidoras de HbF o yuxtaponen los promotores de γ-globina a regiones potenciadoras remotas. Varios estudios demostraron que BCL11A, un represor transcripcional, es necesario para mantener el silenciamiento de la expresión de HbF. BCL11A interactúa con GATA1, FOG1, SOX6 y el complejo represor NuRD y ocupa sitios críticos dentro del grupo de genes de β-globina, incluidas secuencias específicamente eliminadas en individuos HPFH. Estas observaciones proporcionan una fuerte justificación para desarrollar la terapia génica y la edición del genoma enfoques para el tratamiento de las β-hemoglobinopatías destinadas a inducir una conmutación inversa de la globina β a γ.
Referencias bibliográficas
ok 1
ResponderBorrar